Accelerare la scoperta di nuovi farmaci con modelli generativi di diffusione.
Speeding up drug discovery with generative diffusion models.
I ricercatori del MIT hanno creato DiffDock, un modello che potrebbe un giorno essere in grado di trovare nuovi farmaci più velocemente rispetto ai metodi tradizionali e ridurre il potenziale per gli effetti collaterali avversi.
Con il rilascio di piattaforme come DALL-E 2 e Midjourney, i modelli generativi di diffusione hanno raggiunto una popolarità diffusa, grazie alla loro capacità di generare una serie di immagini assurde, mozzafiato e spesso degne di meme a partire da prompt di testo come “orsetti di peluche che lavorano su una nuova ricerca sull’AI sulla luna negli anni ’80”. Ma un team di ricercatori presso la Clinica Abdul Latif Jameel per l’apprendimento automatico in campo sanitario del MIT (Jameel Clinic) pensa che ci potrebbe essere di più nei modelli generativi di diffusione che creano solo immagini surreali: potrebbero accelerare lo sviluppo di nuovi farmaci e ridurre la probabilità di effetti collaterali avversi.
Un articolo che presenta questo nuovo modello di docking molecolare, chiamato DiffDock, verrà presentato all’11ª Conferenza Internazionale sulle Rappresentazioni di Apprendimento. L’approccio unico del modello al design computazionale dei farmaci rappresenta un cambiamento di paradigma rispetto agli strumenti all’avanguardia attualmente utilizzati dalla maggior parte delle aziende farmaceutiche, offrendo una grande opportunità per una revisione della tradizionale pipeline di sviluppo dei farmaci.
- Un sistema robotico a quattro zampe per giocare a calcio su vari terreni.
- I ricercatori del MIT CSAIL discutono le frontiere dell’AI generativa.
- Il sistema di AI può generare proteine innovative che soddisfano obiettivi di progettazione strutturale.
I farmaci di solito funzionano interagendo con le proteine che compongono il nostro corpo, o con le proteine di batteri e virus. Il docking molecolare è stato sviluppato per ottenere informazioni su queste interazioni prevedendo le coordinate atomiche 3D con cui un legante (cioè una molecola di farmaco) e una proteina potrebbero legarsi insieme.
Sebbene il docking molecolare abbia portato all’identificazione di farmaci che ora trattano l’HIV e il cancro, con ogni farmaco che richiede in media un decennio di sviluppo e il 90% dei candidati farmaco che falliscono costosi trial clinici (la maggior parte degli studi stima che i costi medi di sviluppo dei farmaci siano di circa 1 miliardo a oltre 2 miliardi di dollari per farmaco), non sorprende che i ricercatori stiano cercando modi più rapidi ed efficienti per selezionare le molecole potenziali dei farmaci.
Attualmente, la maggior parte degli strumenti di docking molecolare utilizzati per il design dei farmaci in silico adotta un approccio “sampling e scoring”, cercando una “pose” di legante che si adatti meglio alla tasca della proteina. Questo processo laborioso valuta un gran numero di pose diverse, poi le valuta in base a quanto bene il legante si lega alla proteina.
In soluzioni di deep learning precedenti, il docking molecolare viene trattato come un problema di regressione. In altre parole, “si presume che si abbia un singolo obiettivo che si cerca di ottimizzare e che ci sia una singola risposta corretta”, dice Gabriele Corso, coautore e studente di dottorato al secondo anno del MIT in ingegneria elettrica e informatica che è affiliato al Laboratorio di Scienze Informatiche e Intelligenza Artificiale del MIT (CSAIL). “Con la modellizzazione generativa, si presume che esista una distribuzione di possibili risposte – questo è cruciale in presenza di incertezza”.
“Invece di una singola previsione come in precedenza, ora si consentono più pose da prevedere e ciascuna con una diversa probabilità”, aggiunge Hannes Stärk, coautore e studente di dottorato al primo anno del MIT in ingegneria elettrica e informatica che è affiliato al Laboratorio di Scienze Informatiche e Intelligenza Artificiale del MIT (CSAIL). Di conseguenza, il modello non deve compromettersi nel tentativo di arrivare a una singola conclusione, il che può essere una ricetta per il fallimento.
Per capire come funzionano i modelli generativi di diffusione, è utile spiegarli in base ai modelli di diffusione che generano immagini. Qui, i modelli di diffusione aggiungono gradualmente rumore casuale a un’immagine 2D attraverso una serie di passaggi, distruggendo i dati nell’immagine fino a quando non diventa altro che un rumore granuloso. Una rete neurale viene quindi addestrata a recuperare l’immagine originale invertendo questo processo di rumore. Il modello può quindi generare nuovi dati partendo da una configurazione casuale ed eliminando iterativamente il rumore.
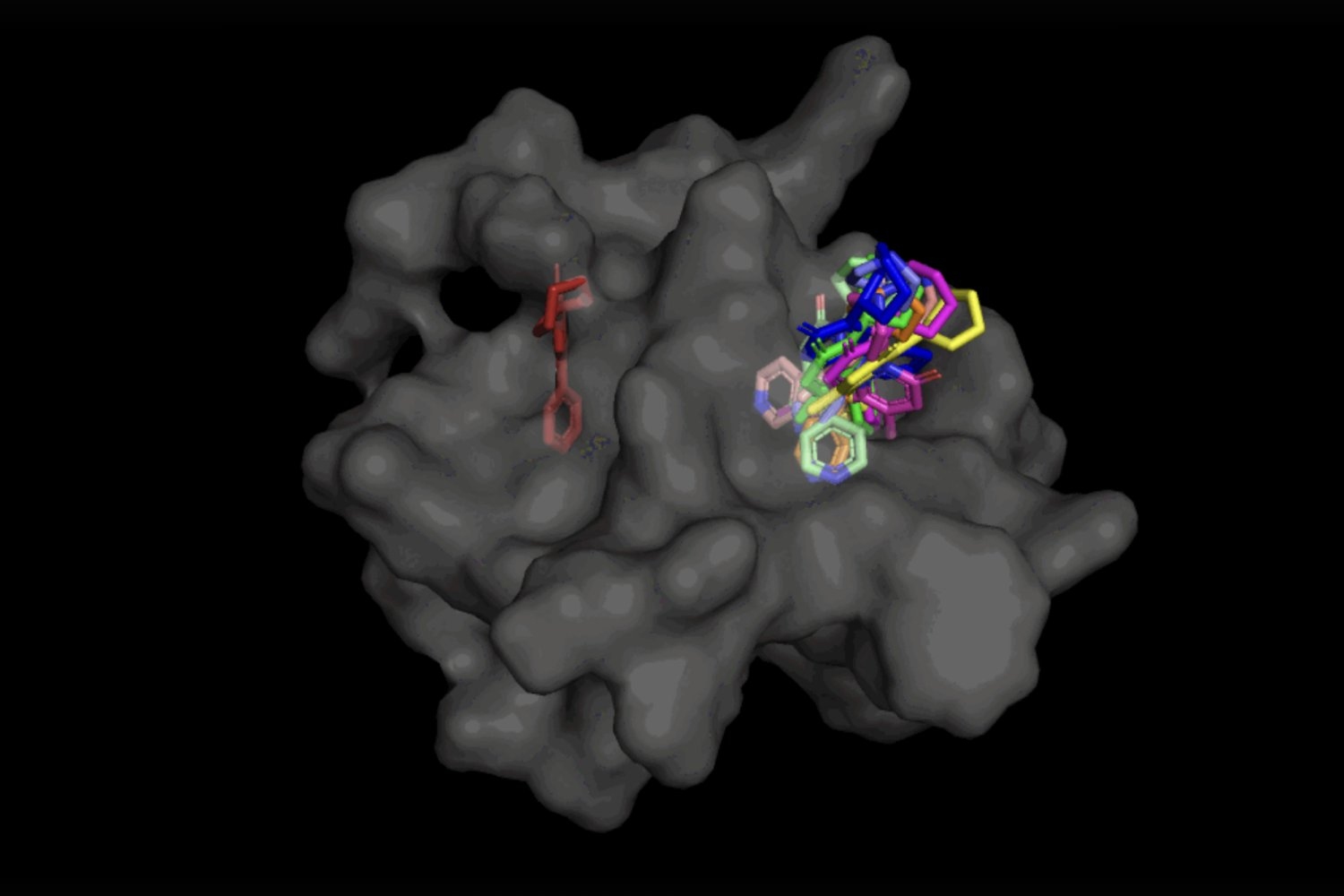
Nel caso di DiffDock, dopo essere stato addestrato su una varietà di pose di leganti e proteine, il modello è in grado di identificare con successo più siti di legame su proteine che non ha mai incontrato prima. Invece di generare nuovi dati di immagine, genera nuove coordinate 3D che aiutano il legante a trovare angoli potenziali che consentirebbero di inserirsi nella tasca della proteina.
Questo approccio di “blind docking” crea nuove opportunità per sfruttare AlphaFold 2 (2020), il famoso modello di AI di piegatura delle proteine di DeepMind. Dall’uscita iniziale di AlphaFold 1 nel 2018, c’è stata molta eccitazione nella comunità di ricerca sul potenziale delle strutture di proteine piegate computazionalmente di AlphaFold per aiutare a identificare nuovi meccanismi d’azione dei farmaci. Ma gli strumenti di docking molecolare all’avanguardia devono ancora dimostrare che le loro prestazioni nel legare i leganti alle strutture computazionalmente previste delle proteine siano migliori di quelle casuali.
Non solo DiffDock è significativamente più accurato rispetto agli approcci precedenti ai benchmark di docking tradizionale, grazie alla sua capacità di ragionare su una scala più elevata e di modellizzare implicitamente parte della flessibilità delle proteine, ma mantiene elevate prestazioni, anche quando altri modelli di docking iniziano a fallire. Nello scenario più realistico che coinvolge l’uso di strutture di proteine non vincolate generate computazionalmente, DiffDock posiziona il 22% delle sue previsioni entro 2 angstrom (considerato ampiamente la soglia per una posa accurata, 1Å corrisponde a uno su 10 miliardi di metri), più del doppio degli altri modelli di docking che si attestano appena sopra il 10% per alcuni e scendono fino al 1,7%.
Questi miglioramenti creano un nuovo panorama di opportunità per la ricerca biologica e la scoperta di farmaci. Ad esempio, molti farmaci vengono scoperti tramite un processo noto come screening fenotipico, in cui i ricercatori osservano gli effetti di un determinato farmaco su una malattia senza sapere su quali proteine il farmaco stia agendo. Scoprire il meccanismo d’azione del farmaco è quindi fondamentale per capire come il farmaco possa essere migliorato e i suoi potenziali effetti collaterali. Questo processo, noto come “screening inverso”, può essere estremamente sfidante e costoso, ma una combinazione di tecniche di piegatura delle proteine e DiffDock potrebbe consentire di eseguire gran parte del processo in silico, consentendo di identificare potenziali effetti collaterali “fuori bersaglio” prima che si svolgano i trial clinici.
“DiffDock rende l’identificazione del bersaglio del farmaco molto più possibile. Prima, era necessario fare esperimenti laboriosi e costosi (mesi o anni) con ogni proteina per definire l’aggancio del farmaco. Ma ora, si possono analizzare molte proteine e fare il triage virtualmente in un giorno”, afferma Tim Peterson, professore associato presso la Scuola di Medicina dell’Università di Washington St. Louis. Peterson ha utilizzato DiffDock per caratterizzare il meccanismo d’azione di un nuovo candidato farmaco per la cura delle malattie legate all’invecchiamento in un recente articolo. “C’è un aspetto molto ‘il destino ama l’ironia’ nel fatto che la legge di Eroom – che la scoperta di farmaci richiede sempre più tempo e costa sempre più denaro ogni anno – sia risolta dalla legge di Moore – che i computer diventano sempre più veloci e meno costosi ogni anno – utilizzando strumenti come DiffDock”.
Questo lavoro è stato condotto dagli studenti di dottorato del MIT Gabriele Corso, Hannes Stärk e Bowen Jing e dai loro supervisori, la Professoressa Regina Barzilay e il Professor Tommi Jaakkola, e è stato supportato dal consorzio Machine Learning for Pharmaceutical Discovery and Synthesis, dalla Jameel Clinic, dal programma DTRA Discovery of Medical Countermeasures Against New and Emerging Threats, dal programma DARPA Accelerated Molecular Discovery, dalla sovvenzione Sanofi Computational Antibody Design e da una borsa di studio del Dipartimento dell’Energia per la laurea in scienze computazionali.






