Da Bit a Biologia #1 Utilizzo dell’algoritmo LCS per l’allineamento globale delle sequenze in Biologia Computazionale
Bit to Biology #1 Using the LCS algorithm for global sequence alignment in Computational Biology.
Un’analisi step-by-step del problema della Sottosequenza Comune Più Lunga e del problema dell’Allineamento delle Sequenze con spiegazione delle loro soluzioni mettendo in evidenza le loro somiglianze e differenze
Introduzione
Benvenuto! Hai trovato il primo articolo della mia serie “From Bits to Biology”, dove esploro le connessioni tra gli algoritmi generali che potresti trovare in una classe di informatica e la biologia computazionale/bioinformatica per rendere tutto più intuitivo e accessibile a coloro che non hanno una formazione formale in biologia. Mi ci è voluto un po’ per convincermi che non è necessario avere una formazione formale in biologia per fare contributi significativi nella biologia computazionale, un campo estremamente affascinante, e spero di convincerti a provarci se ti trovi in una situazione simile 🙂
In questo articolo, stiamo esaminando l’Allineamento delle Sequenze, uno dei problemi più fondamentali della biologia computazionale. L’allineamento delle sequenze di DNA, RNA e proteine ha diverse implicazioni profonde, tra cui la comprensione delle relazioni evolutive, l’avanzamento dell’annotazione funzionale, l’identificazione delle mutazioni e il progresso nella progettazione di farmaci e nella medicina di precisione.
Indice
- Sottosequenza Comune Più Lunga (LCS) – Panoramica LCS – Soluzione di programmazione dinamica LCS – Implementazione in Python di LCS
- Allineamento Globale delle Sequenze (GSA) – Panoramica GSA – Sistemi di punteggio – Soluzione di programmazione dinamica GSA – Implementazione in Python di GSA
- LCS vs. Allineamento Globale delle Sequenze: Somiglianze e differenze – Tabella riassuntiva
Problema della Sottosequenza Comune Più Lunga (LCS)
La Sottosequenza Comune Più Lunga (LCS) è un classico problema di informatica che identifica la sottosequenza più lunga tra tutte le sottosequenze valide condivise tra 2 o più sequenze di input.
Prima di tutto, chiariremo l’enunciato del problema LCS evidenziando una distinzione importante tra una sottosequenza e una sottostringa. Prendi la sequenza "ABCDE":
- Demistificazione del Machine Learning Librerie e strumenti popolari di ML
- Le prospettive dell’IA nella migrazione del cloud
- Come ho creato l’arte generativa con Python che 10000 crediti DALL-E non potevano comprare
- Una sottostringa è costituita da caratteri contigui nel loro ordine originale. Ad esempio, le sottostringhe valide includono
"ABC",“CD”, ecc., ma non“ABDE”o“CBA”. - Una sottosequenza, d’altra parte, non deve essere contigua ma deve comunque essere nell’ordine originale. Ad esempio, le sottosequenze valide includono
“AC”e“BFG”.
Quindi, la LCS è la più lunga tra tutte le possibili sottosequenze tra 2 o più sequenze. Ecco un esempio rapido:

Nella pratica, la LCS ha molte applicazioni che coinvolgono dati di testo molto più grandi, come la rilevazione di plagio¹ o il comando diff di Unix, che richiedono un’efficiente e affidabile soluzione algoritmica scalabile.
Formalizzazione della soluzione LCS
Ecco un ripasso della soluzione di programmazione dinamica a 3 fasi per LCS:
- Inizializzazione (crea una matrice m × n, dove m e n sono le lunghezze della sequenza 1 e delle sequenze 2).
- Riempi la matrice secondo la formula di ricorrenza data di seguito.
- Trova la LCS e la sua lunghezza attraverso il traceback.
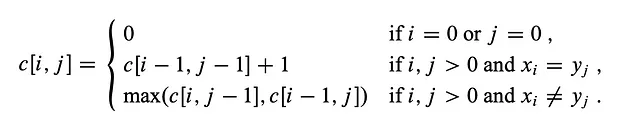
Considera gli input “LONGEST” e “STONE”. Sulla base della relazione di ricorrenza data sopra, riempiamo la matrice DP e poi facciamo un traceback come segue:
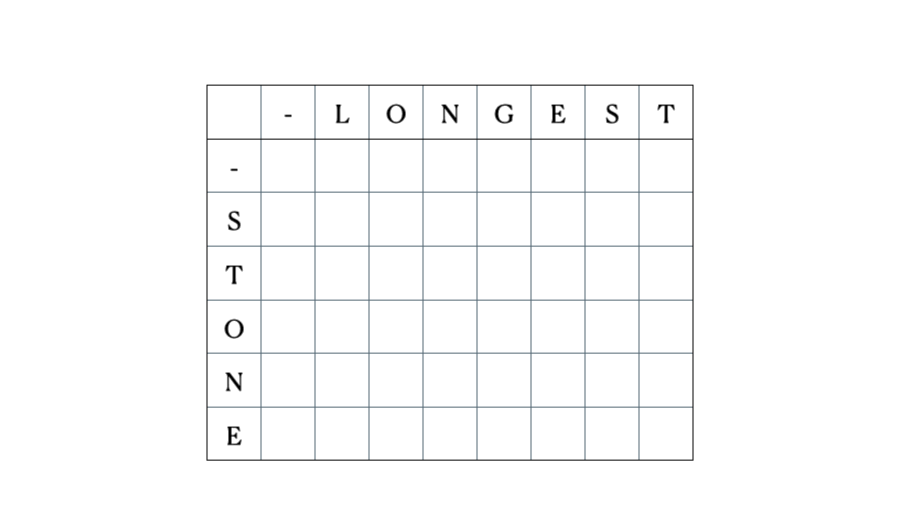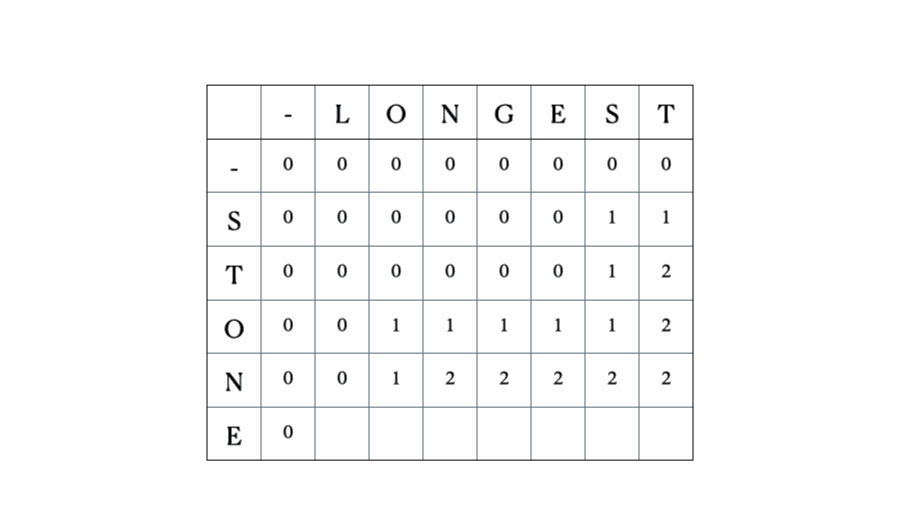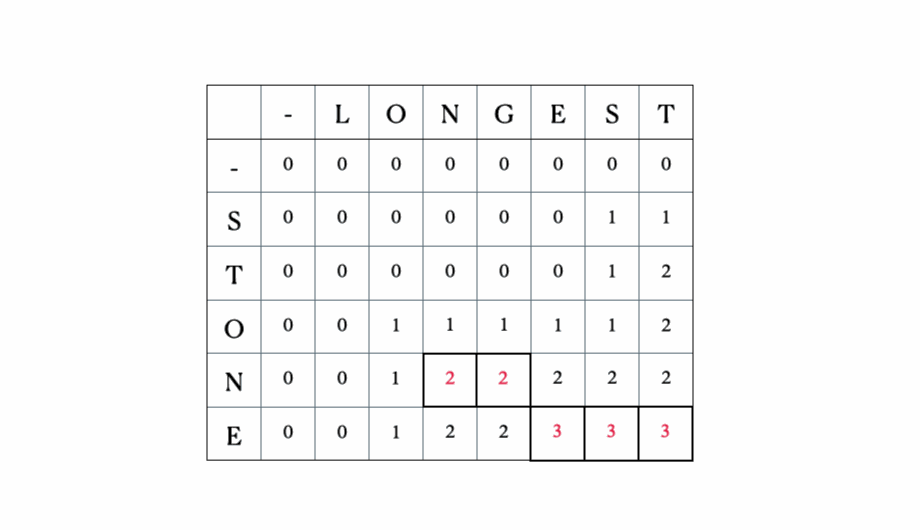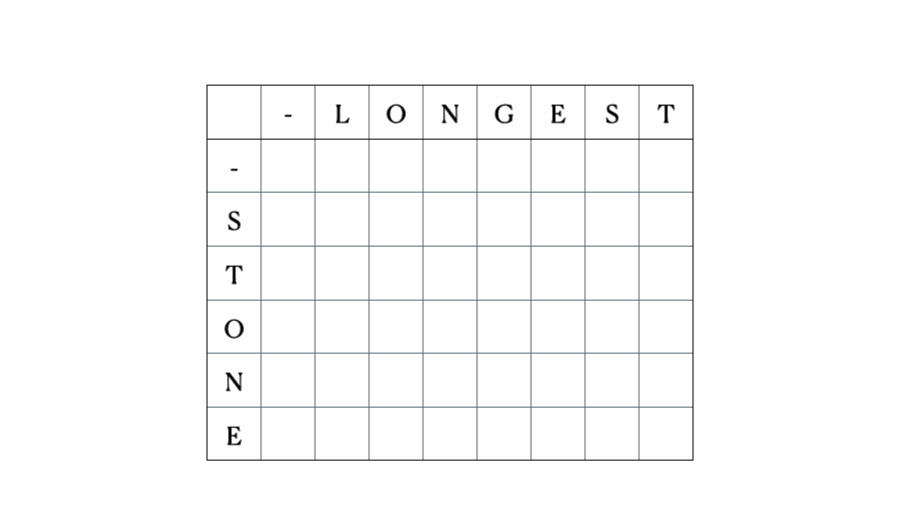
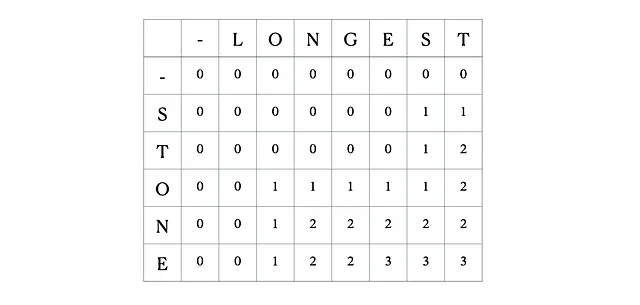
Questa è la tabella DP completa:
Questo si traduce nella seguente implementazione Python:
# Sottosequenza Comune Più Lungadef lcs(seq1, seq2): m = len(seq1) n = len(seq2) # Costruzione della tabella dp table = [[0 for x in range(n+1)] for x in range(m+1)] # Riempimento della tabella sulla base della formula ricorsiva (15.9) di CLRS for i in range(m+1): for j in range(n+1): if i == 0 or j == 0: # caso 1 table[i][j] = 0 elif seq1[i-1] == seq2[j-1]: # caso 2 table[i][j] = table[i-1][j-1] + 1 else: # caso 3 table[i][j] = max(table[i-1][j], table[i][j-1]) # Inizio del traceback dalla cella in basso a destra index = table[m][n] lcs = [""] * (index+1) lcs[index] = "" i = m j = n while i > 0 and j > 0: if seq1[i-1] == seq2[j-1]: # aggiungi i caratteri corrispondenti a LCS lcs[index-1] = seq1[i-1] i -= 1 j -= 1 index -= 1 elif table[i-1][j] > table[i][j-1]: i -= 1 else: j -= 1 # Stampa la sottosequenza comune più lunga e la sua lunghezza print("Sequenza 1: " + seq1 + "\nSequenza 2: " + seq2) print("LCS: " + lcs + ", lunghezza: " + str(len(lcs)))Esempio di utilizzo:
seq1 = 'STONE'seq2 = 'LONGEST'lcs(seq1, seq2)Output:
Sequenza 1: STONESequenza 2: LONGESTLCS: ONE, lunghezza: 3Ora, diamo un’occhiata a come questo algoritmo viene utilizzato nel… ↓
(Problema di) Allineamento Globale di Sequenze
Perché vogliamo allineare sequenze di DNA/RNA/proteine?
In termini intuitivi, allineare due sequenze significa individuare segmenti identici tra le due sequenze, servendo l’obiettivo finale di valutare il livello di similarità tra di esse. Relativamente semplice concettualmente, questa attività è sempre stata piuttosto complessa nella pratica a causa dei numerosi cambiamenti che possono verificarsi nelle sequenze di DNA e proteine attraverso l’evoluzione. In particolare, esempi comuni di questi cambiamenti includono:
- Mutazione puntuale, attraverso inserimento, cancellazione o sostituzione:- Esempio di inserimento: AAA → ACAA- Esempio di cancellazione: AAA → AA- Esempio di sostituzione: AAA → AGA
- Fusione di sequenze o segmenti da geni diversi
Questi cambiamenti indesiderati possono introdurre ambiguità e differenze che oscurano la nostra comprensione di una data sequenza. È qui che utilizziamo tecniche di allineamento per valutare la similarità tra sequenze e inferire caratteristiche come l’omologia.
Come allineiamo le sequenze?
A causa di questi cambiamenti evolutivi e della natura dei confronti tra sequenze biologiche, a differenza di LCS, dove semplicemente scartiamo gli elementi “non abbinate” e manteniamo solo il segmento comune, nell’allineamento globale di sequenze, dobbiamo allineare tutti i caratteri, il che significa che gli allineamenti spesso coinvolgono 3 componenti:
- Corrispondenze, indicate da
|tra le basi corrispondenti - Divergenze, indicate da spazi vuoti tra le basi non corrispondenti
- Inserzioni di gap, indicate da trattini
—
Ecco un esempio. Supponiamo di avere due sequenze di DNA “ACCCTG” e “ACCTGC”. Ci sono diverse modalità per allinearle base per base, tenendo conto delle 3 operazioni di cui abbiamo parlato in precedenza:
Potremmo allinearle senza alcuna inserzione di gap:
ACCCTGT||| |ACCTGCTQuesta allineamento ha 0 inserzioni di gap, 3 corrispondenze, seguite da 3 divergenze e poi 1 corrispondenza alla fine.
O potremmo consentire inserzioni di gap in cambio di più corrispondenze:
ACCCTG-T||| || |ACC-TGCTQuesto allineamento ha 6 corrispondenze totali, 2 gap e 0 divergenze (non contiamo una coppia base-gap come divergenza, poiché una penalità per gap è già inclusa nell’inserzione di gap).
O, se lo desiderassimo davvero, potremmo allinearli anche in questo modo, ottenendo solo 1 corrispondenza totale:
-ACC-CT-GT | ACCTG-CT--Tra qualsiasi coppia di sequenze, quasi sempre ci sono infinite modalità per generare allineamenti tecnicamente corretti come abbiamo appena visto. Alcuni di essi saranno chiaramente peggiori di altri, ma ci saranno alcuni che sono “approssimativamente ugualmente buoni”, come le opzioni 1 e 2 sopra.
Quindi, come valutiamo la qualità di ogni allineamento, scegliamo l’allineamento ottimale e risolviamo le situazioni di parità se necessario?
Schemi di punteggio
1. ACCCTGT 2. ACCCTG-T 3. -ACC-CTGT ||| | ||| || | | ACCTGCT ACC-TGCT ACCTG-CT-Nell’esempio che abbiamo visto in precedenza, ogni allineamento coinvolge una combinazione diversa di numeri di corrispondenze, divergenze e gap.
A differenza del problema della LCS dove una sottosequenza viene valutata solo sulla base della sua lunghezza – più lunga è meglio -, nell’allineamento delle sequenze spesso misuriamo la qualità di un allineamento definendo un sistema di punteggio quantitativo che comprende i 3 componenti chiave di ogni accoppiamento base per base:
- Ricompensa per la corrispondenza: il punteggio aggiunto al punteggio totale dell’allineamento ogni volta che c’è un accoppiamento base per base nella nostra sequenza.
- Penalità per gap: il valore di penalità sottratto dal punteggio totale dell’allineamento ogni volta che introduciamo un gap.
- Penalità per divergenza: il valore di penalità sottratto dal punteggio totale dell’allineamento ogni volta che abbiamo una divergenza.
Come formalizziamo tutto ciò in un algoritmo?
La soluzione algoritmica si sovrappone significativamente alla soluzione della LCS in termini di struttura. È anche un processo in 3 fasi, noto come l’algoritmo di Needleman-Wunsch:
- Inizializzazione (creare una matrice m × n, dove m e n sono le lunghezze della sequenza 1 e delle sequenze 2)
- Riempire la matrice secondo la formula ricorsiva data di seguito
- Tracciare all’indietro per trovare l’allineamento ottimale così come la sua lunghezza
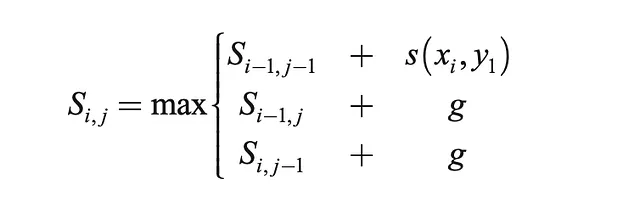
Prima di arrivare alla formulazione matematica e Python della soluzione, ecco una rapida illustrazione del processo con le sequenze “ACGTGTCAG” e “ATGCTAG”:
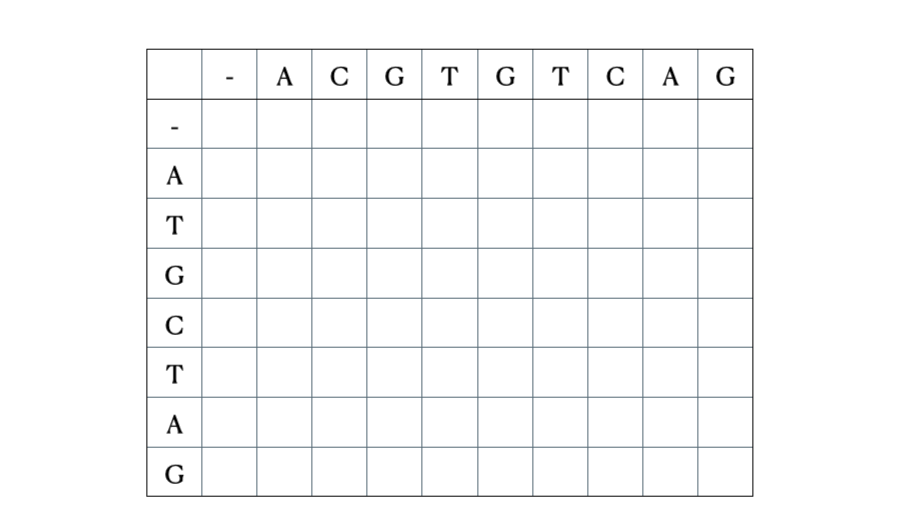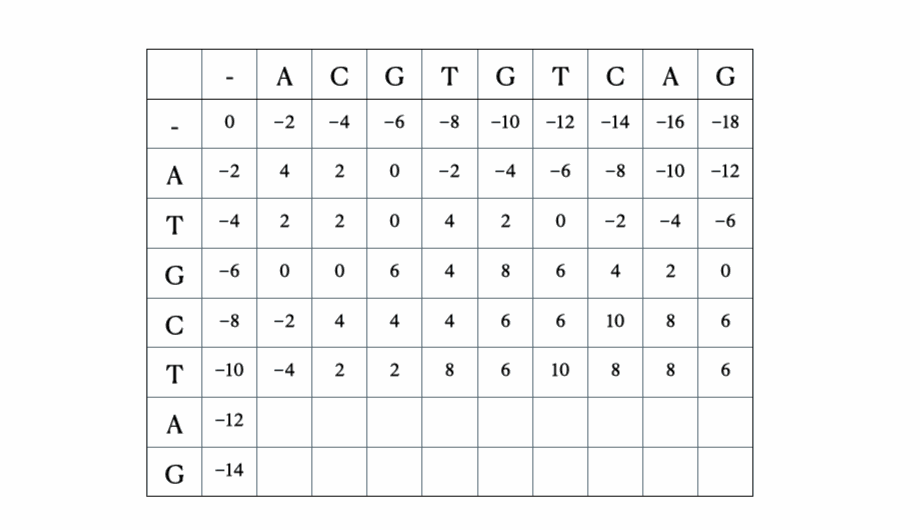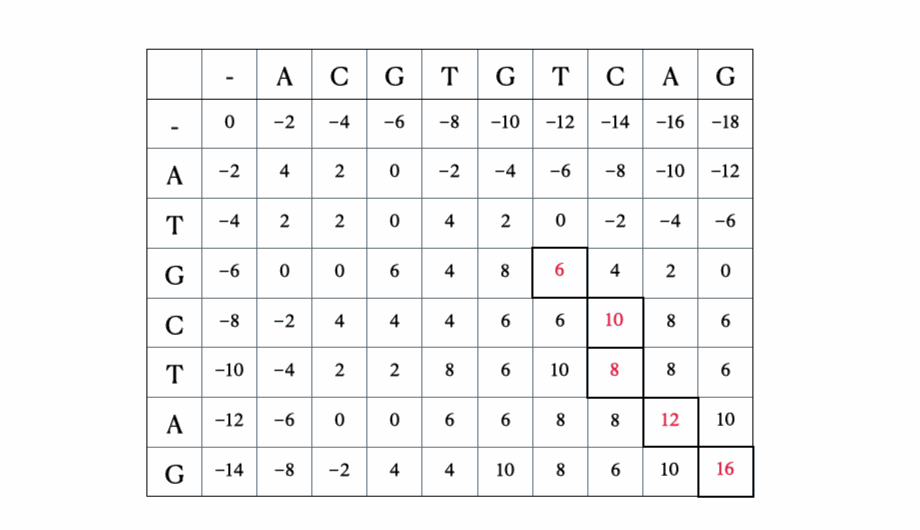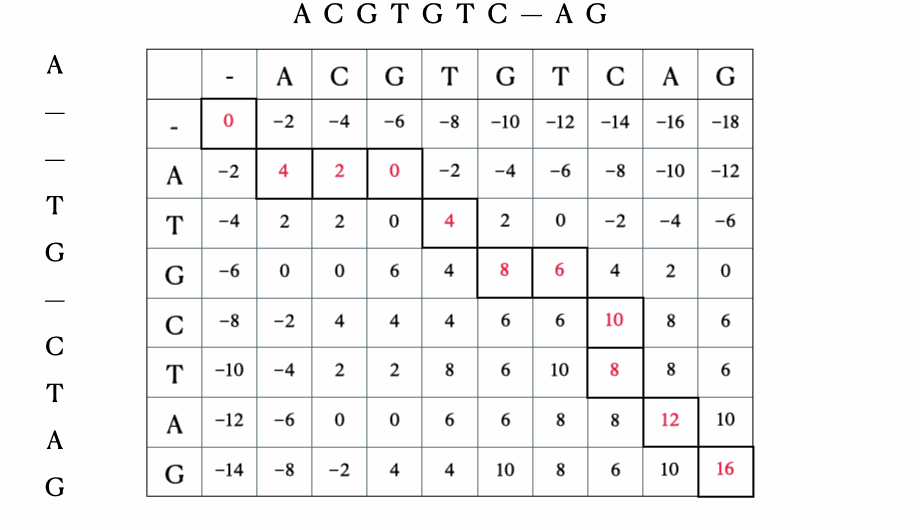
Soluzione in Python
Implementiamo la soluzione in Python utilizzando una classe, GlobalSeqAlign, che gestisce sia la compilazione della matrice che il traceback.
Prima di tutto, costruiamo un oggetto che contiene le 5 informazioni chiave: sequenza 1, sequenza 2, punteggio di match, penalità di mismatch e penalità di gap.
class globalSeqAlign: def __init__(self, s1, s2, M, m, g): self.s1 = s1 self.s2 = s2 self.M = M self.m = m self.g = gSuccessivamente, definiamo una funzione ausiliaria che restituisce il punteggio tra due basi a seconda che siano un match, un mismatch o una coppia base-gap:
def base_pair_score(self, char1, char2): if char1 == char2: return self.M elif char1 == '-' or char2 == '-': return self.g else: return self.mOra, definiamo una funzione molto simile alla funzione LCS che abbiamo visto in precedenza, che utilizza una matrice m × n, la riempie secondo la formula ricorsiva, e poi traccia indietro per restituire l’allineamento ottimale che massimizza il punteggio totale:
def optimal_alignment(self, s1, s2): m = len(s1) n = len(s2) score = [[0 for x in range(n+1)] for x in range(m+1)] # Calcola la tabella dei punteggi for i in range(m+1): # Inizializza con 0 score[i][0] = self.g * i for j in range(n+1): # Inizializza con 0 score[0][j] = self.g * j for i in range(1, m+1): # Riempie le celle rimanenti in base alla formula ricorsiva sopra for j in range(1, n+1): match = score[i-1][j-1] + self.base_pair_score(s1[j-1], s2[i-1]) delete = score[i-1][j] + self.g insert = score[i][j-1] + self.g score[i][j] = max(match, delete, insert) # Traceback, iniziando dalla cella in basso a destra align1 = "" align2 = "" i = m j = n while i > 0 and j > 0: score_curr = score[i][j] score_diag = score[i-1][j-1] score_up = score[i][j-1] score_left = score[i-1][j] if score_curr == score_diag + self.base_pair_score(s1[j-1], s2[i-1]): # match align1 += s1[j-1] align2 += s2[i-1] i -= 1 j -= 1 elif score_curr == score_up + self.g: # verso l'alto align1 += s1[j-1] align2 += '-' j -= 1 elif score_curr == score_left + self.g: # a sinistra align1 += '-' align2 += s2[i-1] i -= 1 while j > 0: align1 += s1[j-1] align2 += '-' j -= 1 while i > 0: align1 += '-' align2 += s2[i-1] i -= 1 return(align1[::-1], align2[::-1]) # Ordine inversoUna volta ottenuto l’allineamento ottimale, possiamo procedere con l’analisi per quantificare quanto bene le sequenze corrispondono.
Quantificazione della similarità: similarità percentuale
Uno dei modi più semplici per misurare il “livello di similarità” è attraverso la similarità percentuale. Prendiamo l’allineamento ottimale, guardiamo la sua lunghezza e la dividiamo per la lunghezza totale delle regioni corrispondenti.
Ad esempio:
Seq 1: ACACAGTCAT |||| |||||Seq 2: ACACTGTCATCi sono 9 corrispondenze su un totale di 10 coppie, il che corrisponde a una similarità del 90%. Ci sono molti altri modi interessanti per analizzare la qualità di un allineamento, ma il meccanismo sottostante dell’allineamento delle sequenze rimane lo stesso.
Confronto tra LCS e Allineamento globale delle sequenze
Ora che abbiamo esaminato dettagliatamente entrambi i problemi e le loro soluzioni, diamo uno sguardo al LCS e all’allineamento globale delle sequenze fianco a fianco.
Similitudini
- Obiettivo: entrambi i problemi cercano e massimizzano le similitudini e gli elementi comuni nelle sequenze di input.
- Approccio algoritmico: entrambi i problemi possono essere risolti utilizzando la programmazione dinamica, che prevede una matrice/tabella DP, il riempimento della matrice e il tracciamento per produrre le sequenze/allineamenti.
- Sistema di punteggio: entrambi i problemi hanno punteggi associati a corrispondenze, non corrispondenze (e spazi) nella corrispondenza delle sequenze. Questi punteggi vengono utilizzati esplicitamente e implicitamente nell’algoritmo per recuperare la sottosequenza/allineamento ottimale.
Differenze
- Sistemi di punteggio: LCS adotta automaticamente un sistema di punteggio di corrispondenza = 1, non corrispondenza/spazio = 0; l’allineamento globale delle sequenze ha un sistema di punteggio più complesso definito da punteggio di corrispondenza = A, penalità di non corrispondenza = B, penalità di spazio = C.
- Gestione degli spazi: LCS di solito tratta gli spazi come non corrispondenze perché per definizione di sottosequenze, vengono conservate solo le corrispondenze; l’allineamento globale delle sequenze assegna di solito un punteggio di penalità agli spazi, diverso dalla penalità di non corrispondenza se c’è una preferenza per la non corrispondenza/spazio.
- Lunghezza dell’allineamento: LCS si preoccupa di trovare un segmento comune a tutti gli input, ignorando gli elementi che non fanno parte di questo segmento; l’allineamento globale delle sequenze corrisponde alle intere sequenze date, anche se significa inserire spazi se le sequenze di input differiscono in lunghezza.
Ecco una tabella riassuntiva dei punti sopra:

Conclusioni
Grazie per avermi seguito fin qui 😼! In questo articolo, ci siamo concentrati su un problema specifico di programmazione dinamica: la Sottosequenza Comune più Lunga (LCS), e abbiamo fatto un confronto diretto con la sua applicazione in biologia computazionale per l’Allineamento delle Sequenze.
In seguito in questa serie, esamineremo anche altri argomenti, come i Modelli Nascosti di Markov, i Modelli a Miscele Gaussiane e gli algoritmi di Classificazione, e come risolvono problemi specifici nella biologia computazionale. A presto e buon coding ✌🏼!
[1] Baba, K., Nakatoh, T., & Minami, T. (2017). Plagiarism detection using document similarity based on distributed representation. Procedia Computer Science, 111, 382–387. https://doi.org/10.1016/j.procs.2017.06.038
[2] Cormen, T. H. (Ed.). (2009). Introduction to algorithms (3rd ed). MIT Press.
[3] Zvelebil, M. J., & Baum, J. O. (2008). Understanding bioinformatics. Garland Science.




